Partitioning large data sets (50m+ reads)¶
“Partitioning” is what khmer calls the process of separating reads that do not connect to each other into different logical bins. The goal of partitioning is to apply divide & conquer to the process of metagenomic assembly.
Basic partitioning¶
The basic workflow for partitioning is in the figure below:
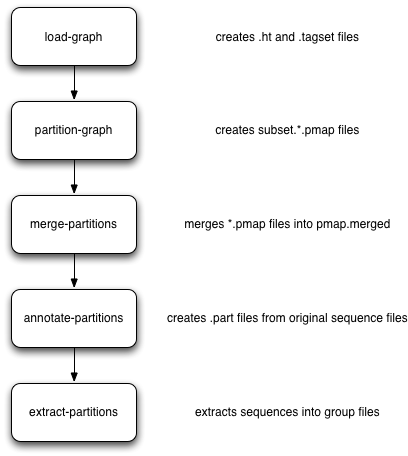
Briefly, you load everything into khmer’s probabilistic graph representation; exhaustively explore the graph to find all disconnected sequences; merge the results of the (parallelized) graph exploration; annotate sequences with their partition; and then extract the different partitions into files grouped by partition size. These groups can then be assembled individually.
Artifact removal¶
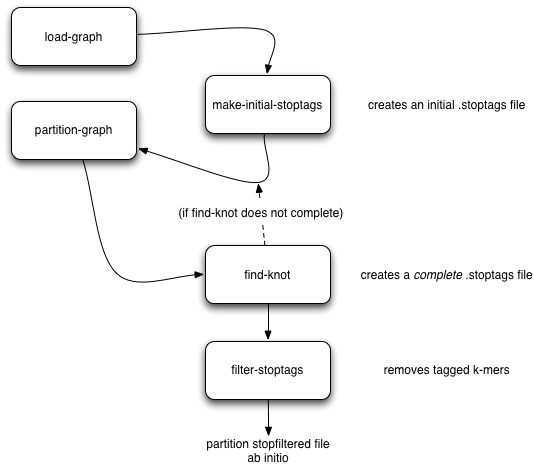
As part of our partitioning research, we discovered that large Illumina data sets tend to contain a single large, connected component. This connected component seems to stem from sequencing artifacts that causes knots in the assembly graph. We have developed tools to forcibly remove the knot at the heart of the graph.
Here’s the workflow:
Running on an example data set¶
Here is a set of commands for running both basic partitioning and artifact removal on a small soil metagenomics data set that we’ve made available for this purpose.
The data set is about 1.1G and you can download it from here:
https://s3.amazonaws.com/public.ged.msu.edu/khmer/iowa-corn-50m.fa.gz
cd /path/to/data
# the next command will create a '50m.ct' and a '50m.tagset',
# representing the de Bruijn graph
load-graph.py -k 32 -N 4 -x 16e9 50m iowa-corn-50m.fa.gz
# this will then partition that graph. should take a while.
# update threads to something higher if you have more cores.
# this creates a bunch of files, 50m.subset.*.pmap
partition-graph.py --threads 4 -s 1e5 50m
# now, merge the pmap files into one big pmap file, 50m.pmap.merged
merge-partitions.py 50m
# next, annotate the original sequences with their partition numbers.
# this will create iowa-corn-50m.fa.gz.part
annotate-partitions.py 50m iowa-corn-50m.fa.gz
# now, extract the partitions in groups into 'iowa-corn-50m.groupNNNN.fa'
extract-partitions.py iowa-corn-50m iowa-corn-50m.fa.gz.part
# at this point, you can assemble the group files individually. Note,
# however, that the last one them is quite big? this is because it's
# the lump! yay!
# if you want to break up the lump, go through the partitioning bit
# on the group file, but this time with a twist:
mv iowa-corn-50m.group0005.fa corn-50m.lump.fa
# create graph,
load-graph.py -x 8e9 lump corn-50m.lump.fa
# create an initial set of stoptags to help in knot-traversal; otherwise,
# partitioning and knot-traversal (which is systematic) is really expensive.
make-initial-stoptags.py lump
# now partition the graph, using the stoptags file
partition-graph.py --stoptags lump.stoptags lump
# use the partitioned subsets to find the k-mers that nucleate the lump
find-knots.py -x 2e8 -N 4 lump
# remove those k-mers from the fasta files
filter-stoptags.py *.stoptags corn-50m.lump.fa
# now, reload the filtered data set in and partition again.
# NOTE: 'load-graph.py' uses the file extension to determine
# if the file is formatted as FASTA or FASTQ. The default is
# fasta, therefore if your files are fastq formatted you need
# to append 'fastq' to the name so that 'load-graph.py'
# will parse the file correctly
load-graph.py -x 8e9 lumpfilt corn-50m.lump.fa.stopfilt
partition-graph.py -T 4 lumpfilt
merge-partitions.py lumpfilt
annotate-partitions.py lumpfilt corn-50m.lump.fa.stopfilt
extract-partitions.py corn-50m-lump corn-50m.lump.fa.stopfilt.part
# and voila, after all that, you should now have your de-knotted lump in
# corn-50m-lump.group*.fa. The *.group????.fa files can now be
# assembled individually by your favorite assembler.
Post-partitioning assembly¶
The ‘extract-partitions’ script takes reads belonging to each partition and aggregates them into ‘group’ files; each group file contains at least one entire partition (and generally a lot more). Note, you can control the number of reads in each file (equiv, the size of these files) with some of the arguments that ‘extract-partitions’ takes.
Now that you have these files... what do you do with them? The short answer is: assemble them! Each of these group files contains reads that do not connect to reads in other files, so the files can be assembled individually (which is the whole point of partitioning).
If you’re using Velvet, checkout the sandbox/velvet-assemble.sh script, which you can run like this:
bash /path/to/khmer/sandbox/velvet-assemble.sh <groupfile> <k>
This script does three things:
- first, it breaks the reads up into paired reads and single reads, and puts them in separate files (.pe and .se);
- second, it strips off the partition information from the reads, which confuses Velvet;
- and third, it runs velveth and velvetg to actually assemble.
You can implement your own approach, of course, but this is an example of what we do ourselves.